Advanced Tutorial#
In this tutorial, we will show how to model a ferromagnetic CoO system and include a Hubbard U correction to improve the description of the electronic structure in highly correlated systems like transition metal oxides. Finally, you will also learn how to set up a simulation for an antiferromagnetic configuration (but this time, without a Hubbard U).

You can define the initial magnetic moments for each chemical element in your structure. It is also possible to set different initial magnetic moments to inequivalent sites of a given chemical element, see the antiferromagnetic example. Moreover, you can also define the U values and set them specifically for each chemical element.#
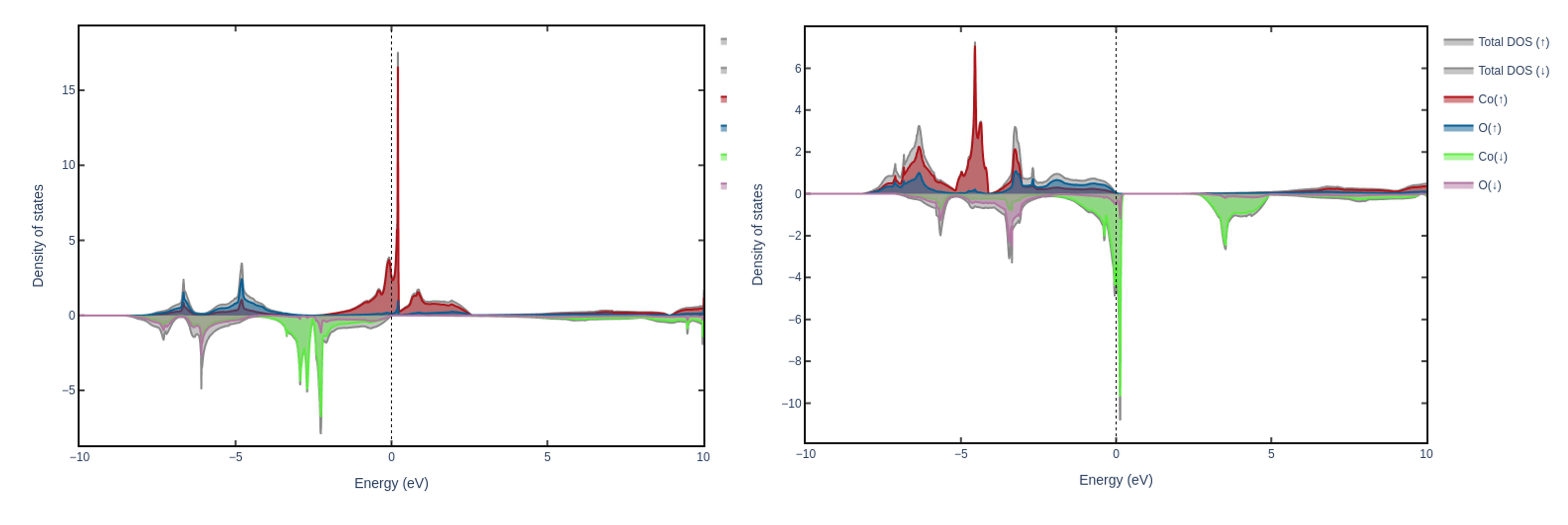
The left plot shows the PDOS of CoO without the inclusion of a Hubbard U correction. On the contrary, the right plot presents the PDOS after assigning the Hubbard U of 5 eV to the Co-3d states, which leads to the redistribution of these states.#

To model an antiferromagnetic configuration, you need to set the initial magnetic moments with opposite signs to the two Co atoms in the unit cell. You will learn in the in-app guide how to modify your structure to define the two different types of Co atoms!#
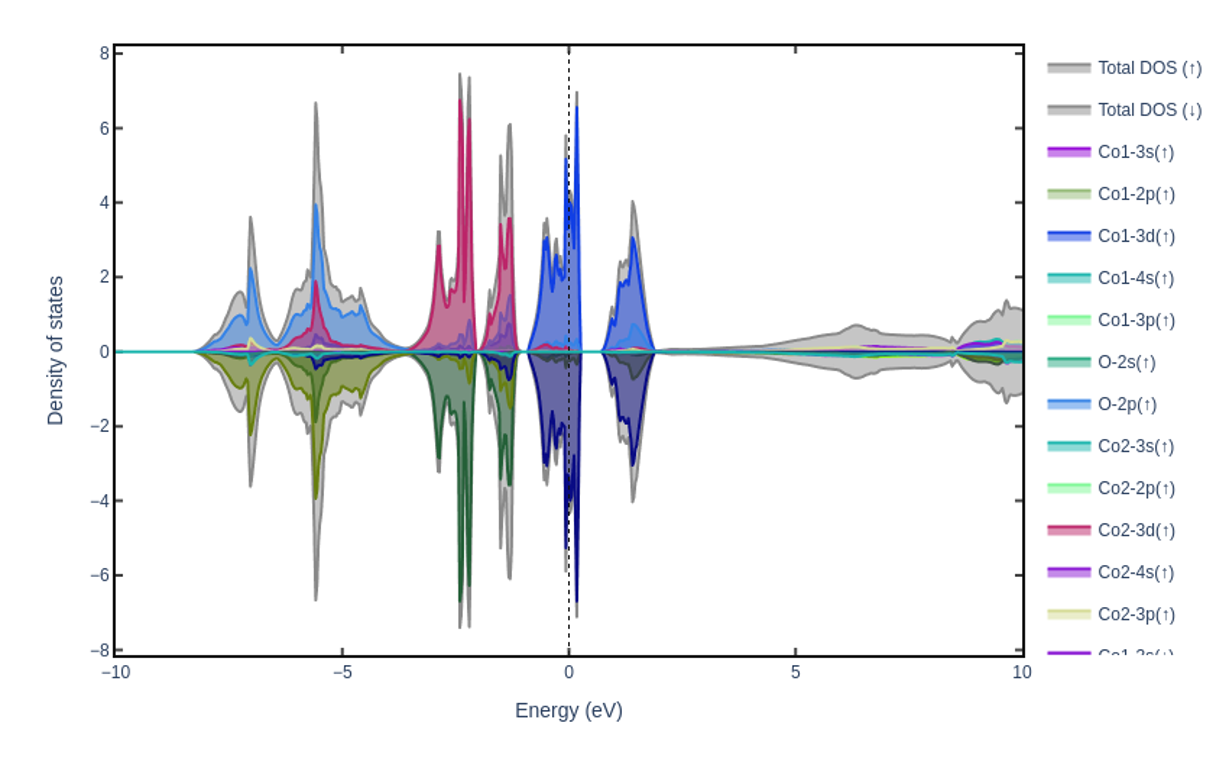
The resulting PDOS for the antiferromagnetic configuration of CoO (without Hubbard U).#